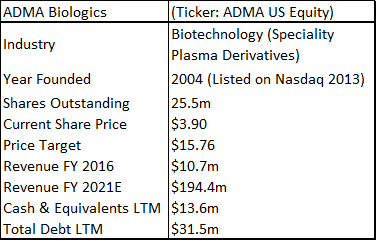
ADMA Biologics: Almost Ready To Run Between The Toes Of Giants
1. Introduction to the Investment Thesis
ADMA Biologics is a sub-$100m capitalization healthcare company that is based in Boca Raton, Florida. The company is aiming to commercialize a novel high-titer hyper-immune globulin product for the treatment of RSV and related respiratory viruses in immune compromised patients. ADMA listed as a public company in February 2013 (on the Nasdaq OTC market through a reverse merger).
The two main contentions of this positive investment thesis are; 1), the burden of lifting the regulatory hold at the Boca Raton facility, while being a significant challenge for management, should prove to be within reach; 2), The long-term potential of entering the plasma market as a small supplier of niche high-value hyperimmune products is a very attractive business proposition for ADMA. While it is unusual for large and strategically important fractionation facilities to experience regulatory issues, the overall regulatory landscape pertaining to plasma processing, fractionation and logistics is clearly defined by the relevant regulatory authorities.
ADMA has had three capital injections since it took control of the Boca facility. It obtained additional financing as part of the BTBU transaction, it refinanced its debt with Marathon Asset Management, and it raised $39.1m in additional equity. ADMA management is now confident that it will have sufficient cash resources to progress the company into the first half of 2018, in advance of key corporate milestones. ADMA's current cash burn rate is c.$3m to c.$3.5m per month, and the company ended Q3 2017 with c.$13m in cash. Revenues for the nine months of 2017 were $10.8m. Through the transaction agreement, ADMA secured a ten-year plasma supply agreement with Biotest AG (and/or its successors), along with a three-year post-closing agreement, for the purchase of RSV plasma from the two collection centers, at a price equal to cost plus 5% (excl-inflation). Biotest AG is currently engaged in a potential buy-out process with Chinese private equity firm Creat Group (an affiliate of Shanghai RAAS, the China based pharmaceutical conglomerate). ADMA appears to be stuck between a rock and a hard place. The company is burning its cash resources and appears to be stuck in regulatory limbo. However, upon detailed analysis it is clear that these concerns are overblown.
If ADMA can pass an FDA cGMP re-inspection at the first time of asking, it will be in a much stronger strategic position relative to when it first became a public company. Currently the market isn't willing to ascribe any 'benefit of the doubt' valuation on the shares, and the company is being valued as an entity with considerable capital but with few valuable assets. From the perspective of the long-term healthcare investor, ADMA shares now represent a very favorable investment opportunity. The company is 'off the radar' for the average investor, but still possesses a relatively liquid share float and has a listing on the Nasdaq. Perhaps the most interesting aspect surrounding ADMA's current valuation, is the lack of value that ADMA's current share price places on RI-002 (especially in light of the current overvaluation in the biotechnology industry). ADMA has generated strong phase III data (in fact the drug produced some of the strongest clinical results ever for an IVIG). The drug has a modest hurdle for approval and there is a significant unmet medical need among immune compromised (IC) patients, who are vulnerable to developing RSV and other common respiratory viruses. ADMA's approach of isolating high-titer antibodies to specific infections is a novel one, with the larger plasma company's focusing on recombinant technologies, hemophilia and breakthrough vaccines for rare diseases.
2. 2018 is the make-or-break year for ADMA
The upcoming FDA re-inspection at the Boca facility is the key business milestone for ADMA. Management has guided that the facility will be ready for re-inspection in late 2017.
Figure 2.1: Anticipated milestones in Q4 2017 and beyond
Source: Analyst Estimates
3. The BTBU transaction has transformed ADMA strategic positioning
The fractionation facility that ADMA has acquired is 126,000 sq.m in size, and has a peak capacity of 400,000L per annum. The site also includes a state-of-the-art monoclonal antibody production facility, along with 15 acres of land adjoining the buildings. The facility employed c.900 employees and generated total revenues of $76.5m in FY2016.
Figure 3.1: The BTBU generated $76.5m in revenues for FY 2016

Source: ADMA Biologics Prem. Form 14A
Biotest originally purchased the Florida facility from Nabi Pharmaceuticals in 2007, (Nabi had built the facility in 2001) for a transaction value of $185m (which incl. 9 plasma collection centers). Biotest invested $50m in upgrading and expanding the facility and invested $70m in constructing an adjoining state-of-the-art monoclonal antibody production unit. The transaction should be highly accretive for new shareholders. With vertical integration, management have guided that a blended gross profit margin ranging c.60% to 70% is achievable (prior guidance was for top-line margins of c.40% to 50%).
4. Primary Immune Deficiency and the adaptive immune system
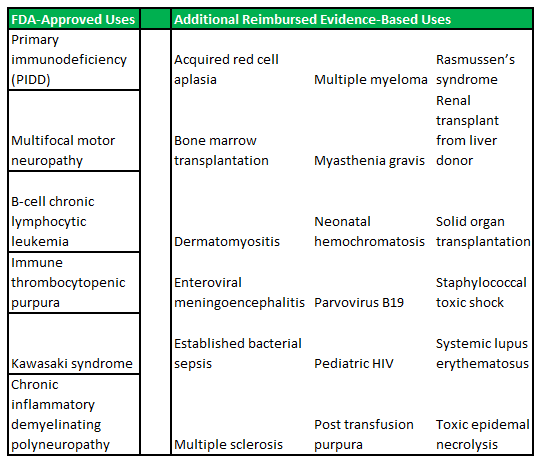
Primary immune deficiency disorder is the collective term for a range of medical disorders that occur when the adaptive immune system is malfunctioning. Patients with one or more forms of PIDD, often have defects in their t-cells or b-cells (important for the immune system's ability to adapt to the presence of foreign pathogens) as opposed to defects in the innate immune system (phagocyte and complement disorders). There are relatively concrete population prevalence estimates, that can be gathered from a range of studies conducted in hospitals across the United States. These place the total US patient population, at approximately 1 in 1,200, or 250,000 people.
PIDD disorders are commonly viewed by physicians as falling into the rare diseases category. The exact prevalence is difficult to estimate, as there are no standard screening procedures for immune system defects in the United States (or elsewhere in the world). The main disorders that fall under the PIDD category are listed in figure 4.1 (below).
Figure 4.1: ADMA intends to focus on the high-risk segments within the PIDD patient population

Source: ADMA Biologics
IVIG has been the global standard of care for PIDD patient's in recent decades, and IVIG treatments are still viewed by physicians as the most appropriate treatment option for PIDD. IVIG's are a powerful cocktail of immune system antibodies, which are isolated from pools of plasma collected from thousands of plasma donors. The concept of immune globulin use has a long history across the medical landscape and has been widely indicated for use in passive immunization. In these instances there is a high risk of infection and the body has insufficient time to develop its own immune response, or to reduce the symptoms of ongoing or immunosuppressive diseases. While the functioning of the immune system is well characterized, it has proven very difficult to replicate the near natural effects of immune system supplementation that can be achieved through IVIG treatment (to date chemical and monoclonal antibody formulations have not achieved similar effects). The increasing use of self-administered solutions, which allow patients to treat their conditions in an out-patient setting, has contributed to the predominant position that IVIG now occupies. IVIG use is particularly beneficial among young children and babies which have under-developed immune systems. Infants have particular safety and efficacy needs which often can't be accommodated through treatment with standard vaccines.
On the other hand, IVIG's have unique treatment profiles. As the formulations are largely comprised of the natural human antibodies, their safety profile is limited to complications that might arise during the manufacturing process, or with complications that might be observed among plasma donors, or with complications that might be observed during the administration of the final product. IVIG's can be combined with other therapies, including new therapies that are introduced onto the market, without having to demonstrate efficacy or drug-drug interactions, etc. IgG can be modified for the use of MaB's and used as a carrier to enhance therapeutic efficacy.
When side effects do result from IVIG use, they have resulted from the tendency of IgG to form aggregates or clots when the plasma is purified during the manufacturing stage. This is often caused by filter clogging. If a patient is administered a solution that contains coagulants it can cause potentially severe reactions. Complications can also arise from excessive donation and this has been a focal point for the anti-donation lobby (more discussion in Section 17 below). The physical effects experienced during a donation session are not dissimilar to those experienced while donating blood, e.g., mild injection site reactions, feelings of nausea, etc., although reaction rates have been reported to be higher for blood donations. The frequency of acute reactions among donors undergoing apheresis procedures was found to be 2.18% in a multi-institutional study. In comparison, in a review of re actions among whole blood donors, an overall reaction rate to whole blood donation of 11 to 21% was reported. A 2005 study found hypocalcemia to be the most common aphaeresis-specific reaction (caused by citrate anticoagulation). Further study is needed to determine the effect this might have on the long-term plasma donor. Hypocalcemia is caused by the excess presence of sodium citrate in the blood stream. This bonds with calcium in the blood, which in turn leads to reduced circulating calcium levels, and in extreme cases can result in hypocalcemia. In non-extreme cases donors can experience fainting, tingling, numbness, muscle contractions or seizures. Bone demineralization can also result from long-term decalcification. In the PIDD subset of IC patients, c.50% of the US population is actively using an IVIG formulation.
RSV has an infection rate of c.5%-15% in the IC population (off-label use is widespread). ADMA contends that physicians will be receptive to using a proven medical solution to treat the full range of IC patients, who are vulnerable to contracting respiratory viruses. It is widely understood that patients are often misdiagnosed and/or left undiagnosed for a number of years after the initial presentation of symptoms. The American Academy of Pediatrics (in its bronchiolitis management guidelines), does not specifically recommend RSV testing. This increases the risk for recurrent upper and lower respiratory tract infections, as well as for bacterial sepsis, which can be immediately fatal.
Reverse transcription polymerase chain reaction is an expensive assay testing technique that is not commonly used outside of the emergency department setting, owing to the expense and lack of available results while the child is in the ED. A study of US children under the age of 5 years who were tested for RSV independent of their discharge diagnosis, showed that only 45% were diagnosed with an RSV-associated illness, while in the outpatient setting RSV positive children had a 3% RSV diagnosis. RSV is commonly misdiagnosed as otitis media, dyspnea, wheezing, and croup.
Patients who continually suffer recurrent respiratory infections are placed at an increased risk of suffering chronic lung disease and bronchiectasis. The benefit of therapeutic immunoglobulin use in this population is seen in its ability to reduce the incidence rate of infections. This has been attributed to the diverse antibody specificities against common viral pathogens present in plasma formulations. While the FDA requires IVIG products to pass potency tests for minimal levels of antibodies against measles, diphtheria, and polio, these pathogens do not commonly result in infections among PIDD patients. Currently available IVIG products are not standardized to contain specific antibody content to defend against the most common infectious diseases that PIDD patients experience. Studies of the available products have reported a wide variation in antibody titres.
When ADMA is able to market its full stable of products, it can focus its sales effort for Bivigam towards the general PIDD population, while RI-002 can be focused towards higher-risk patients. The total addressable market for RI-002 could be c.8,500 to c.15,000 patients. The high-risk/rare disease nature of these patients, should allow RI-002 to command a premium price, compensating for the relatively small total addressable market. ADMA intends to put RI-002 forward for initial approval, with a label indicating treatment for a range of respiratory viruses that are commonly observed among PIDD patients. In light of RI-002's positive phase III results and its strong effect on RSV antibody elevation, it has strong potential to be viewed by physicians as a core treatment option for RSV infection during the winter months (complementary potency against other common respiratory viruses should be viewed very favorably). RI-002 won't be contraindicated with other treatments commonly used by these patients (distinguishing it from vaccines).
Following approval, ADMA can pursue additional indications to treat patients who are vulnerable to contracting RSV (and other common respiratory viruses) due to their IC status. A particularly large group that could benefit from RI-002 treatment, are patients undergoing chemotherapy treatment. These patients are placed at an elevated risk of mortality after contracting viral infections. An important benefit of antibody treatment is that it can be synergistic or additive when combined with conventional antimicrobial chemotherapy against bacterial fungal and viral infections. RI-002 was probably renamed (originally known as RI-001 before management communicated introduction of minor changes in production process) to maintain clinical profile comparability and smooth the process of label expansion post-approval. For plasma derivatives comparability guides the development process during the preclinical phases. This ensures that clinical data is linked to the product intended for commercialization. A robust comparability program during all stages of the product life-cycle shortens the time to licensure and facilitates the implementation of post approval changes. The CBER's plasma products paradigm is that the manufacturing process is the product that is ultimately approved and licensed.
Figure 4.2: IVIG is widely used both on-label and off-label
Source: FDA & CBER.
5. RI-002's phase III results "batted the ball from the park"
RI-002 demonstrated an incidence rate of zero SBI's in 55.88 patient years of treatment, which resulted in an SBI rate below the FDA's criteria of <1.0 SBI per subject per year. Other key secondary endpoints were well within a reasonable range expected to observed in IVIG's (see figure 6.1 below). A difference in specific antibody levels was observed among patients who received doses ≥500 mg/kg in comparison to patients who received doses ≤500 mg/kg.
Figure 5.1: RI-002 demonstrated zero SBI's and a very low rate of hospitalizations due to infection

Source: Wasserman et al.
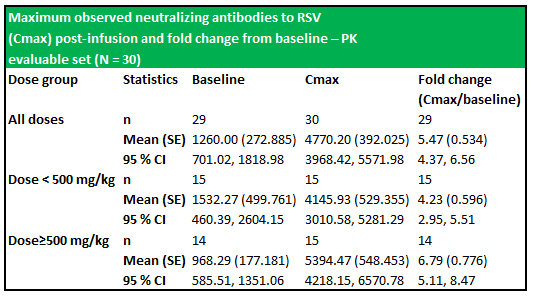
RI-002 administration produced increases in all of the studied antibodies, with the greatest increase observed in antibodies to RSV, with a 5.47-fold increase in titres after infusion of the IVIG and a 6.79-fold increase in those subjects who received greater than 500 mg/kg (figure 6.2 below). The applied baseline measurement was the value of anti-RSV measured immediately prior to trial entry. Figure 6.3 (below) shows the PK of anti-RSV antibodies, where the baseline was the value of anti-RSV measured in subjects enrolled in this portion of the study, immediately prior to infusion of the seventh or ninth dose of IVIG.
Figure 5.2: Mean IgG plasma concentration levels and mean Cmax values were similar across treatment groups despite the slightly longer treatment period

Source: Wasserman et al.
Figure 5.3: The dosage schedule produced significantly elevated levels of RSV antibodies (from baseline) in both treatment groups
Source: Wasserman et al.
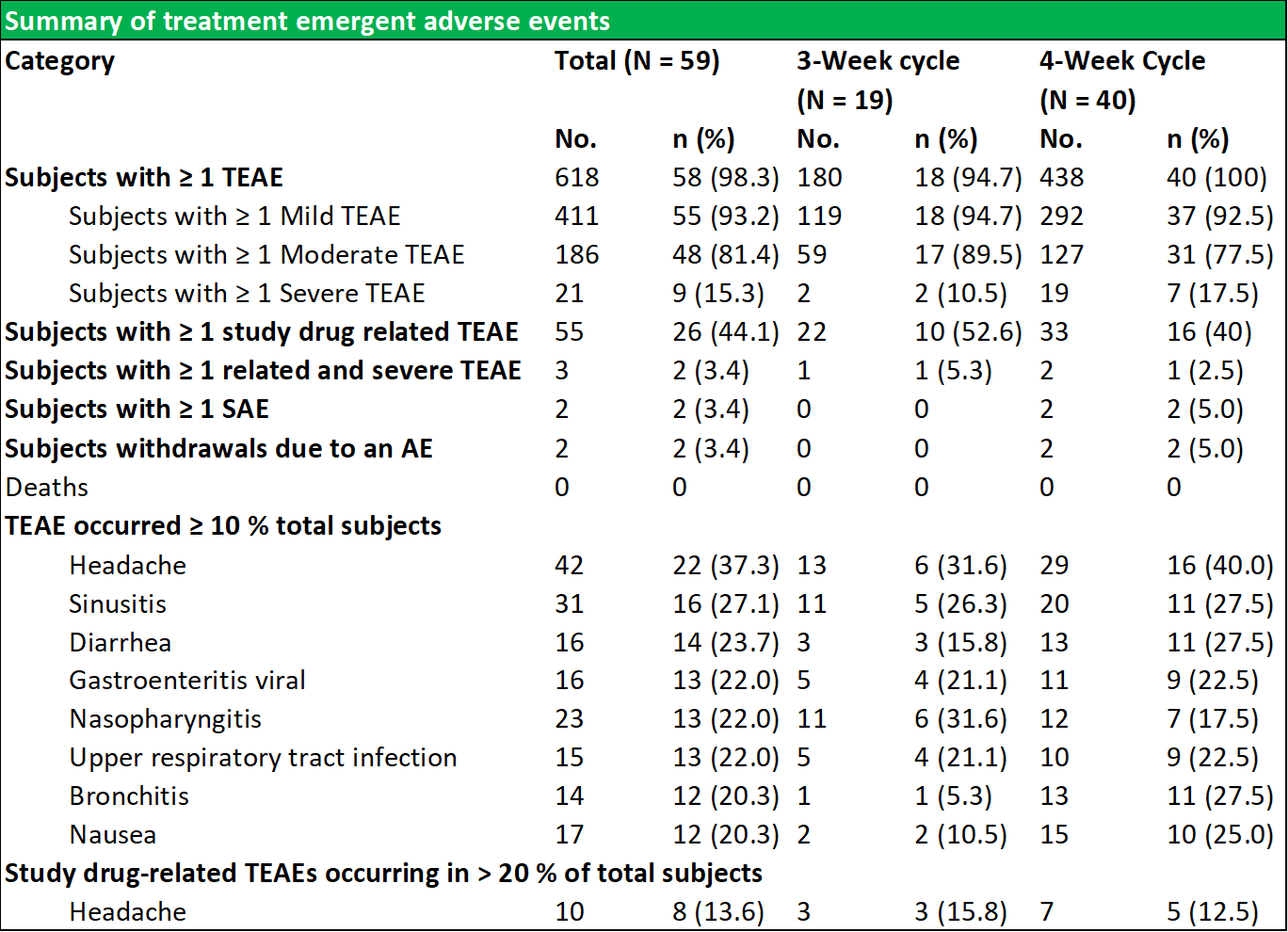
RI-002 was safe and efficacious and the portion of study infusions that experienced a TAAE was 0.142, with an upper one sided 95% confidence limit of 0.164, which was well within the target safety endpoint of (<0.40) (figure 6.4 below documents the adverse events schedules).
Figure 5.4: Summary of temporally associated and treatment emergent adverse events

Source: Wasserman et al.
During the study, 43 (72.9 %) of subjects experienced at least one TAAE, and TAAE's occurred during 113 (14.2 %) of study infusions. The mean (SE) planned weight adjusted administered dose of RI-002 was 505.2 (4.84) mg/kg. Study infusions were well tolerated with 95.8% of infusions administered at the maximum allowed infusion rate of 8 mg/kg/min. The use of pre-medication was limited to seven study infusions (from a total of 793 infusions) in seven subjects (0.009%). Of the 618 TEAE's that were experienced by 58 subjects, the most common were headache, sinusitis diarrhea, viral gastroenteritis, nasopharyngitis, and upper respiratory tract infection (common illnesses in this population). There were no reported serious adverse events related to RI-002, and reported the two events, were experienced as a single event in a single subject.
Figure 5.5: Total IgG PK parameter estimates show well-expressed concentration profiles between subjects in both treatment cycles
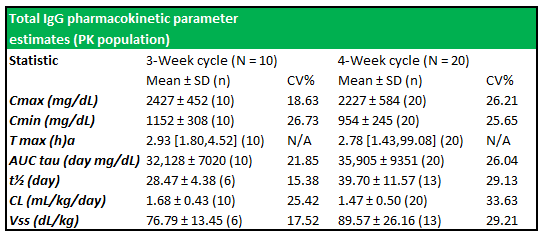
Source: Wasserman et al.
6. ADMA's trip to regulatory purgatory should be short lived
Two important points to consider when assessing the regulatory burden that ADMA faces at the Boca facility; are 1), it is very unusual for fractionation facilities to experience regulatory issues (several industry representatives expressed this view in telephone conversations); 2), the regulatory burden for plasma fractionators is very well defined in the relevant regulatory statutes; 3) industry practices and standards are well-established and there is a considerable industry expert body-of-knowledge in relation to these specific issues. This has built continually built upon over decades of practice in the United States. Speculating as to the reasons why the facility has experienced difficulties over a prolonged period, is not important in the assessment of ADMA's ability to remediate the issues at the facility. ADMA has communicated with investors that the FDA regional inspectors took issue with BTBU responses to questions such as, "what would you do if something went amiss with such-and-such a process". Management outlined that BTBU's provision of clear operating procedures and staff training protocols was below regulatory standards. ADMA feels that if management are more critical of out-of-specification operational and manufacturing processes, and develop procedures that are less cumbersome and easier to follow, they will be able to operate the facility within the regulatory framework. A central aspect of preparing the facility for a re-inspection will be the guidance that it receives from Validation Consulting. Engaging external subject matter experts when changing a manufacturing process is common practice in the fractionation industry. BTBU didn't appear to use this approach when it attempted to change aspects of the processes used to manufacture Bivigam (more details on this aspect below). It is also possible that managing the adjoining MaB bioreactor facility, was an undue burden on BTBU management. Three MaB's were progressed through a clinical development program but were subsequently discontinued.
ADMA has been working closely with BTBU management since the issuance of the CRL in July 2016. This has allowed ADMA to develop a strong understanding of the issues involved. As part of this cooperation period, members of ADMA management along with its Board of Director's toured the Boca Raton facility. This process lead onto the purchase agreement that concluded with BTBU. New staff, new employees, new standard operating procedures (SOP's), new training programs and a comprehensive compliance enhancement program should place the facility in a position to pass a re-inspection. Management has outlined that this program will involve overhauling 16 different quality management systems. Consultants from Validation will provide an Interim Control Plan and act as a third party internal quality oversight function for all production activities. The small team of consultants working at the facility are experts in their field and their fees for Q2 2017 were $2m. ADMA's management has guided that the facility will require minimal additional capital investment. The CRL that ADMA has for RI-002, sets out in detail the various aspects that caused the facility to fail the compliance checks, and the remediation efforts can be focusing on these issues. This is key to removing the valuation overhang on ADMA's share price. At present, the market is unwilling to ascribe any 'benefit of the doubt' valuation on the shares.
Figure 6.1: Over time Investors are hoping that the business climate at the facility will match the local Florida climate



Source: ADMA Biologics
7. ADMA's supply of source plasma is secured from its three existing centers and through the supply agreement with Biotest
Figure 7.1: ADMABiocenters employ c.50 employees and can cater for 50 donors at any one time
Source: ADMA Biologics
ADMA currently has two fully-operational collection centers in Marietta and Norcross (in the State of Georgia), and a third center is due to begin collection activities in January 2018 (in Kennesaw, Georgia). On average it takes one year to obtain FDA approval for a new center, and another operating year to build up a stable donor base. After establishing a stable donor base, it usually takes between two-to-three years before the center has integrated into the local community and a sufficient supply of plasma can be collected to justify the build. The centers advertise in the local community and engage in initiatives designed to increase visibility among the local population. The three centers are c.10,000 sq. feet in size and cost between $2m and $3m to develop. Each center will employ c.50 people and will have peak capacity for 50 donation stations.
Figure 7.3: ADMA's collection centers are worth c.$16m each and can generate a stable revenue stream of c.$6.5m

Source: ADMA Biologics
The BTBU transaction agreement allocated a value of approximately $6.3m for each center. A 2.5x sales multiple is a fair valuation metric for the centers, and a well-run center could be expected to generate approximately $6.5m. While ADMA has agreed to transfer two centers to Biotest AG in 2019, it can benefit from the use of these centers until YE 2019. Importantly, the transaction agreement included a long-term supply agreement with Biotest AG or its successors, which will guarantee ADMA's supply needs.
Figure 7.4: Post transaction ADMA's centers will be c.9 hours by road from the Boca Raton facility. The facility itself is a 12min walk to CSL's Plasma Global headquarters
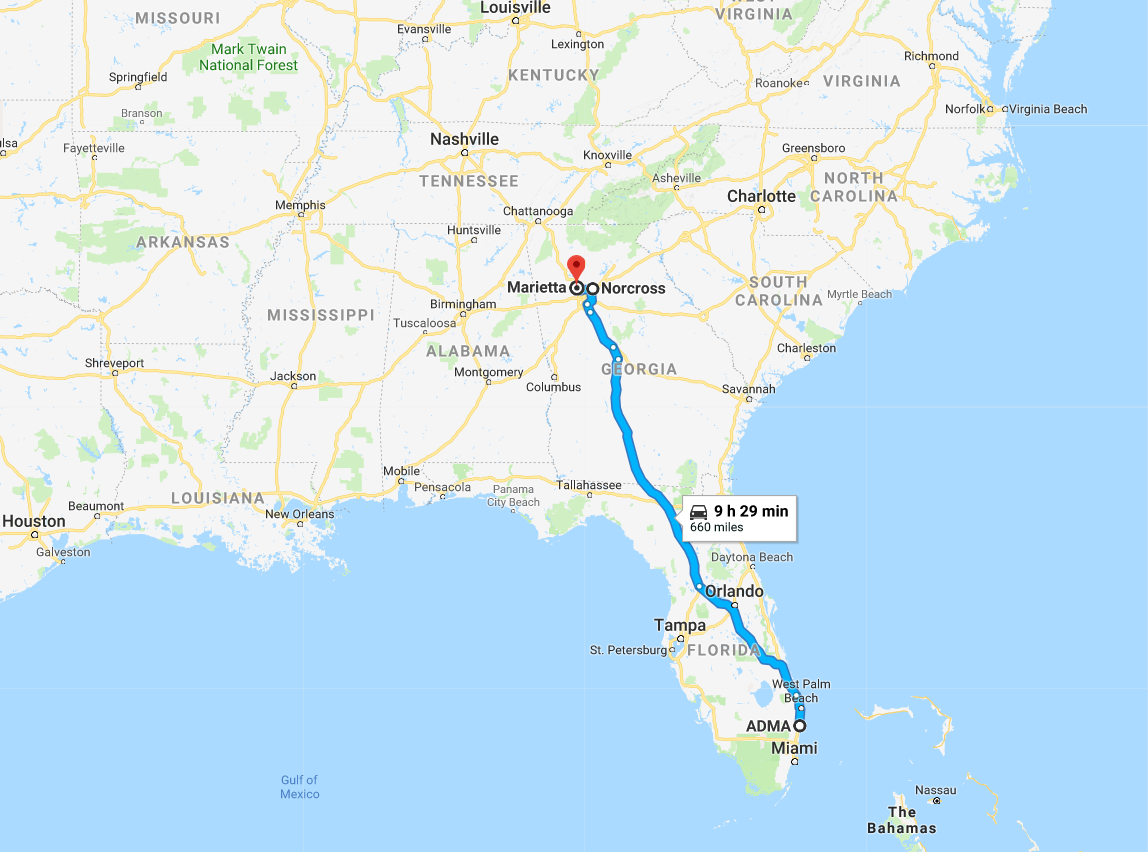
Source: Google Maps
Securing the supply of raw plasma is a key strategic consideration for the large processors. The ability to increase capacity depends in part on the ability of the processor to increase its collection volumes. Developing economies of scale in plasma collection is easier than in plasma fractionation, which requires a higher capital outlay to build new or additional capacity. Historically plasma collection has contributed a greater amount to the overall cost of goods sold, as donor fees are the highest cost in the overall cost base. For the average collection and fractionation operation, collection costs can represent c.50% of the total cost of goods. On a per liter breakdown of cost of goods, the large fractionators are able to achieve a split of c. 30% to 40% for fractionation and c. 60% to 70% for plasma collection.
8. Grifols SA, CSL Ltd and Shire Plc dominate the US plasma collection industry
The number of companies active in the plasma industry has fallen from thirteen companies in 1990 to just six companies at present, while the overall market value size has increased significantly. The total amount of plasma collections has increased from 15.3m in 2007, to 38.3m in 2016, a CAGR of 10.7%. Industry growth is being driven by increasing demand from emerging markets and clinical data which has supported the use of plasma derivatives for additional medical indications. As a small player in a niche segment of a fast-growing market, ADMA has found itself with 'a seat at the table' of a very attractive industry.
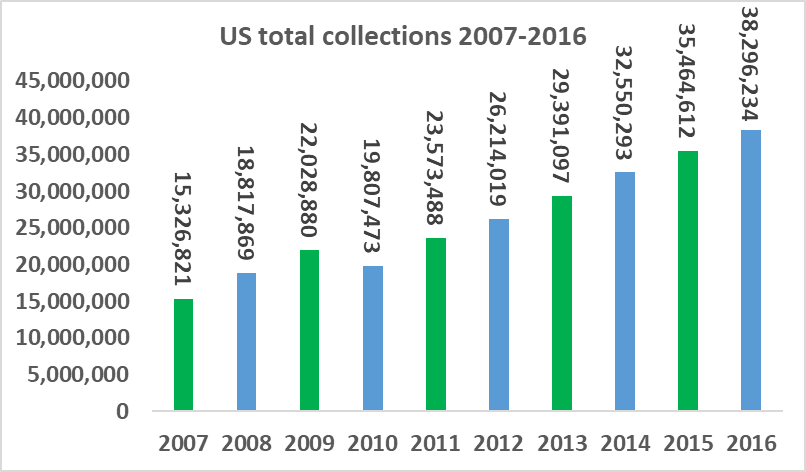
Source: PPTA
This increase in collection capacity allowed for significant growth in the total market value for IVIG, the most valuable plasma derivative. The overall increase in market value was primarily driven by a gradual increase in the price of IVIG. This was driven by the recognition among the medical community that IVIG could be used to treat a broader range of conditions (particularly neurological conditions).
8.1: Average prices for IG rose even as the large players expanded capacity. This dynamic produced significant overall growth in market value
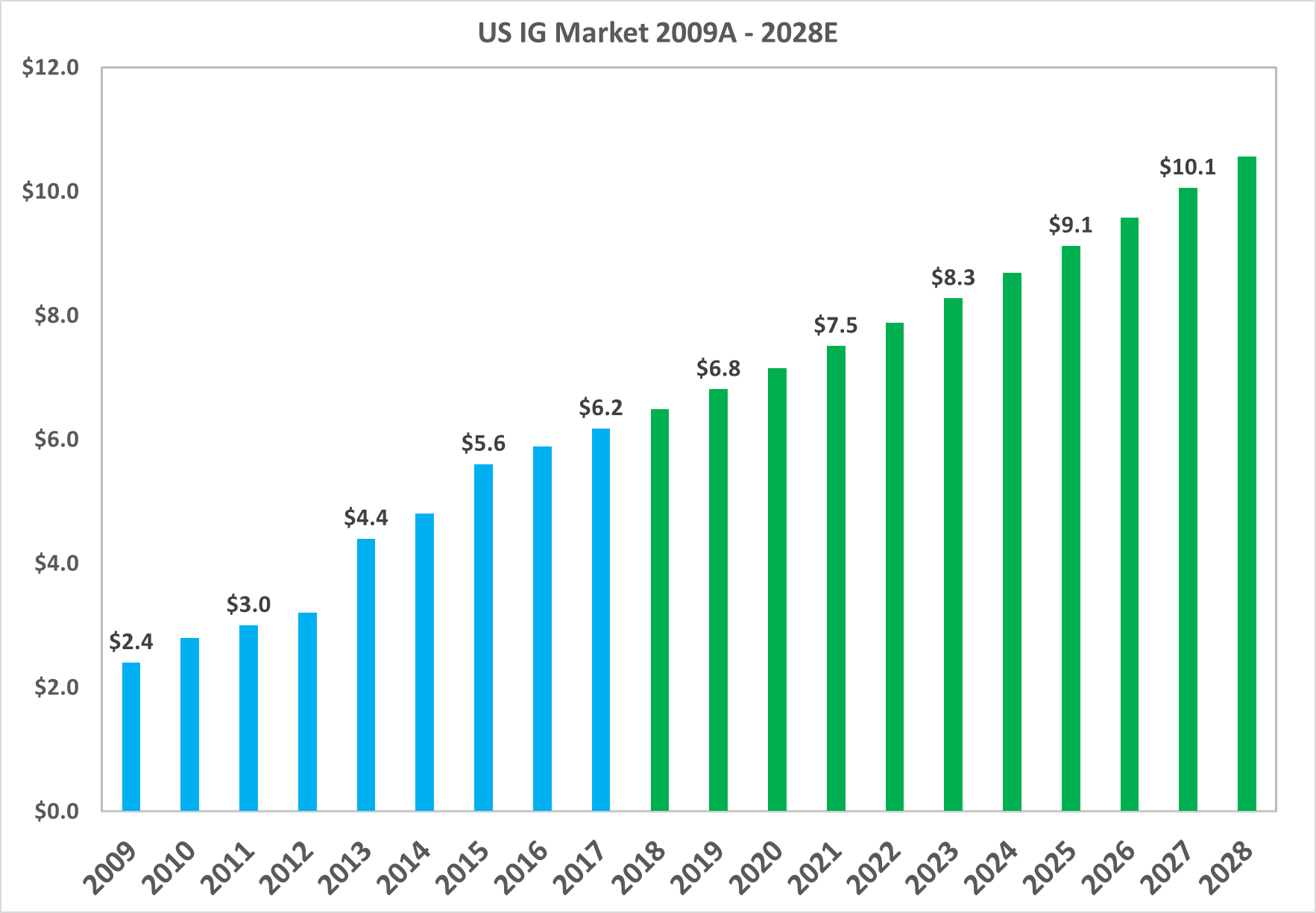
Source: MRB Research, LEK Consulting, Aetna, United Health
The total market value of the North American market grew from $2.4bn in 2009 to $6.2bn in 2017, or a CAGR of 12.2%. The market is projected to continue growing at a CAGR of 5% up to 2028.
8.2. The plasma collection industry is dominated by the three large processors who hold more than 75% market share
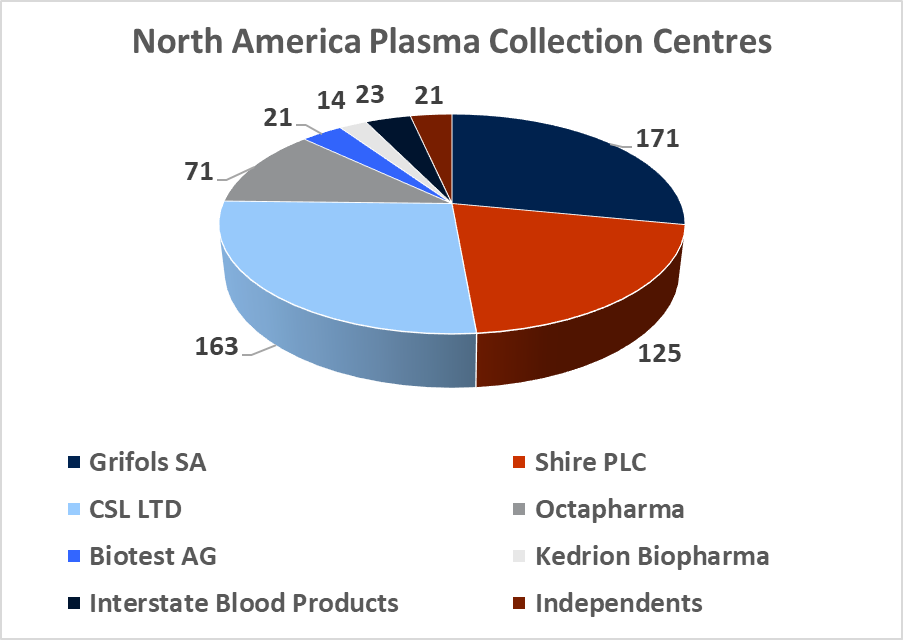
Source: DonatingPlasma.org
At year-end 2017, there was c.670 regulated and fully operational plasma collection centers in the western hemisphere, with 609 located in North America and 57 located in Europe. The total North American plasma collection network grew at a CAGR of 10% between 2012 and 2016 and this growth rate is likely to continue in the coming years (in light of company guidance). The global market leadership position has changed hands several times between Grifols SA and CSL Ltd over the last decade. This depended largely on which company had recently made a large acquisition and thus was able to expand its total processing capacity. In 2017, Grifols SA led the industry with 171 centers, CSL Ltd had 163 centers, Shire Plc (though its $32bn acquisition of Baxalta in 2016) has 125 centers, and Octapharma AG (Private) has 71 collection centers. CSL Ltd has focused on developing its own centers under the CSL Plasma brand, while Grifols SA has pursued a dual expansion strategy, building out new greenfield sites and engaging in transactions with smaller independent collectors. It purchased 49% of Interstate Blood products for $100m in May, 2016 (deal included 9 collection centers), and paid $49m for 6 plasma centers from Kedplasma in February 2017. CSL's global plasma collection operations are concentrated in North America although the company has recently commenced efforts towards building a processing presence in China (purchasing the Rui De plasma business in Wuhan Province for $352m in 2017). The large collectors aim to create clusters of collection centers in the regions where they establish operations with the aim of creating collection efficiency. The two main challenges involved in building out large donor networks (absent the clear regulatory and development burdens) are increasing the donor base and recruiting qualified staff.
8.3: The large plasma processors forced the industry to consolidate by buying up smaller independent networks
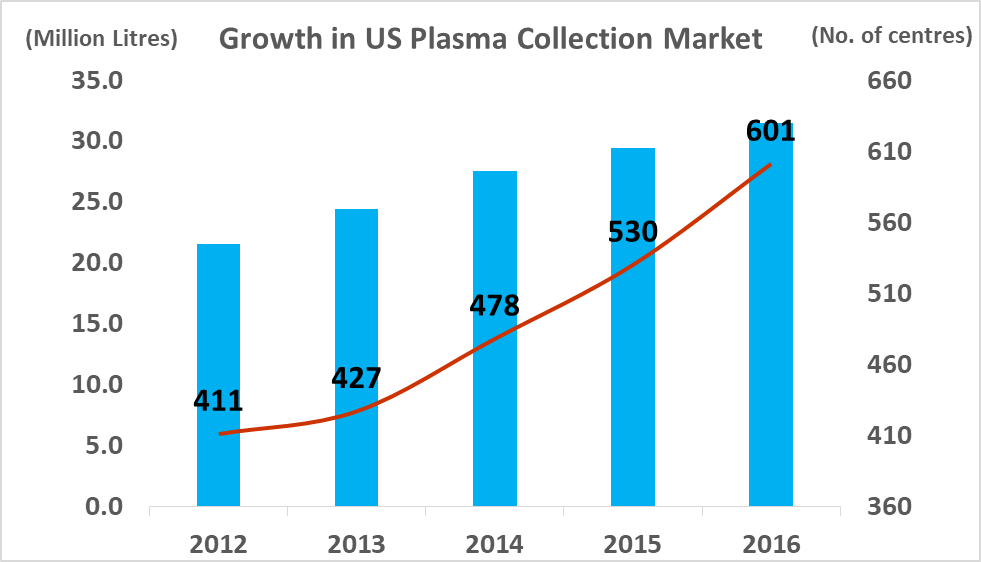
Source: PPTA, DonatingPlasma.org
The structure of the global plasma industry has been crafted through several large transactions, which allowed the purchasers to generate significant economies of scale and achieve substantial processing efficiencies.
In 2000, c.50% of the North American plasma collection market was independently owned, but by 2010 c.80% of the market was held by the three largest companies.
The three dominant companies have conglomerate style organizational structures, which allow them to pursue complimentary business lines in areas such as vaccines, rare diseases and small molecule drug development. Shire achieved significant scale in 2016 through a $32bn purchase of Baxalta (spun off from Baxter Pharmaceuticals in 2015). Baxter had previously sold its fractionation assets to NPS Biotherapeutics (JV by Cerberus Capital Management and Ampersand Ventures). Subsequently Shire Plc has lost the pole position to Grifols and CSL. CSL Ltd began life as a supplier of vaccines in Australia before privatizing and pursing international expansion. In 2003 the company purchased Aventis Behring for $925m and integrated that company's European manufacturing plants into its own operations. Grifols purchased Talecris Biotherapeutics (spun off from Bayer's processing facilities in North Carolina) for $3.4bn in 2010 (acquiring Gamunex, a treatment for three immune-system disorders, and Prolastin, the top-selling treatment for Alpha-1 antitrypsin deficiency, which accounted for 77% of Talecris sales). Grifols SA integrated Talecris' substantial North American collection center network into its existing European processing operations. As a result (and like CSL Ltd) it maintains its own specialized freight service to ship frozen plasma to Europe where it is processed into the plasma derivatives (both companies maintain large processing plants in Europe). Grifols SA, began life as a private blood bank in Spain, before expanding into North America and eventually buying Alpha Therapeutics (from the Japanese company Green Cross) in 2003. Biotest AG began life as a small plasma processor in Germany in the 1860's before gradually expanding into North America. Octapharma was founded in Austria in 1983 as a specialist contract fractionator and today it has significant collection capacity in North America (with 71 centers). Kedrion grew out of a family run blood and plasma business in Italy.
8.4: Grifols SA has outlined details of its long-term capital investment plan
Grifols has given guidance to the market on its planned capacity additions and total planned investment amounts. To at least maintain current relative positioning, CSL Ltd will need to pursue a similar expansion plan. An important aspect of product pricing in the plasma industry is that prices are determined by the cost base of the marginal producer. Therefore, profitability for the large fractionators is tightly linked to relative efficiency.
8.5: The larger processors can expand capacity with incremental EBITDA margin growth

Source: Analyst Estimates
Most industry participants now recognize that the incremental margin growth for all but the most efficient fractionators is close to zero at current market prices. This makes it uneconomical for the smaller fractionators to pursue market growth strategies and it means that the larger fractionators capture an outsized proportion of the year-on-year average growth produced by the industry. As a result, the combined market share of Kedrion, LFB and Biotest AG has declined in recent years, while CSL and Grifolshave increased theirs. Octapharma (private) is the only smaller processor who has been able to profitably expand its capacity. It achieved this through, 1) significant investments in its collection center network in North America (it plans to double output from 2016-2019), 2) aggressive pricing tactics, 3) aggressive expansion into North and South America (Octapharma had lost share after a product recall in 2009). Importantly, Octapharma was able to grow its margins and its top-line revenues in recent years by improving its average fractionation yields and implementing process improvements.
8.6: CSL and Grifols SA will likely increase their positions in the market by matching each other's future growth plans
CSL Ltd likely has the relative efficiency advantage in plasma fractionation at present but Grifols will be attempting to close this gap in the coming years. As part of Grifols' North American capital investment plan ($1.2bn), the company will expand its base of collection centers to 225 by 2019 (from 171 in 2017), and increase its fractionation capacity from 13.9m liters per year to 19.0m liters per year by 2022.
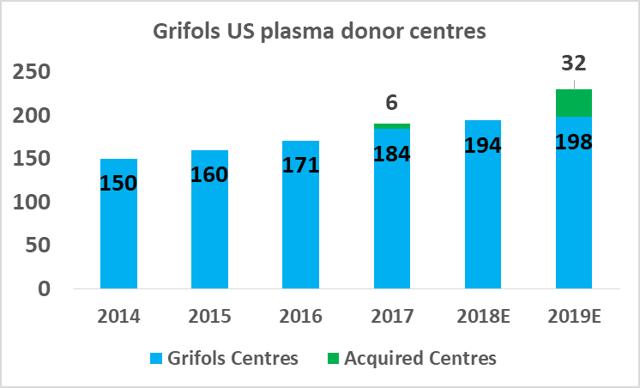
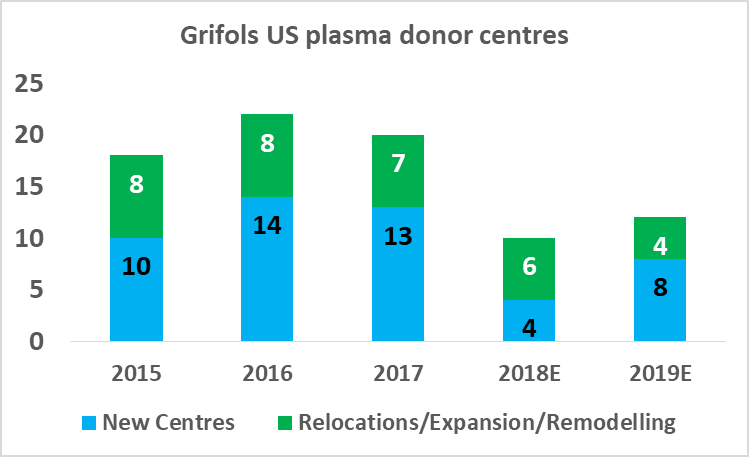

Source: Grifols SA Investor Presentation
8.7: At present, Grifols SA has a slight market share lead in the IVIG segment
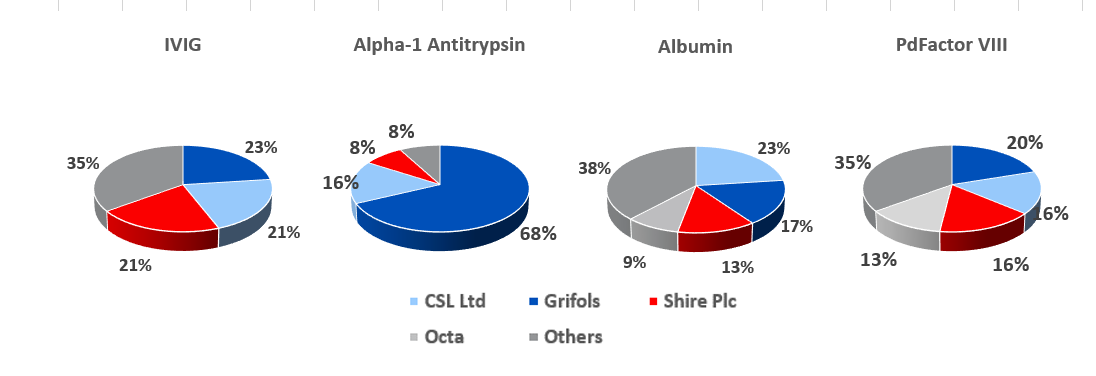
Source: Grifols SA, PPTA
For the big six plasma processors, IVIG is the most profitable protein that can be extracted from each liter of plasma (prices have fluctuated between $60 and $75 per gram in recent years), while albumin is least valuable and yields about 25g per liter of plasma, and currently costs c.$2.50 per gram. The other two most profitable proteins are Alpha-1 Antitrypsin and PdFactor VIII. The economics of plasma fractionation (called last liter economics, is discussed in section 19 below) is such that the fractionator can tailor the various steps involved in its fractionation processes to enhance the yield of a given plasma batch depending on end market supply and demand conditions.
8.8: This Grifols presentation slide shows how an integrated plasma processor structures its business models
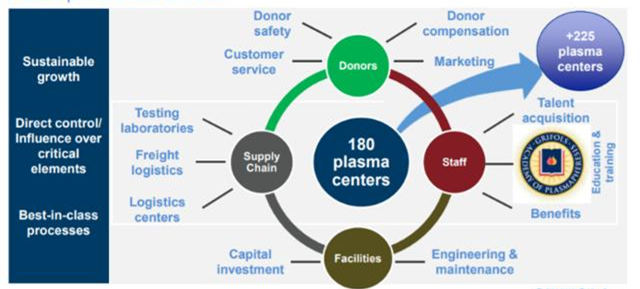
Source: Grifols SA
8.9: The State of Florida is a hub for the plasma industry in the South-Eastern United States
It would be very economically challenging for a small processor like ADMA to enter the traditional plasma market with just two collection centers. Using Grifols as the benchmark for the efficient processor (it discloses significant details on its capacity levels), it can be estimated that c.12 fully-operational collection centers are required to fractionate c.1m liters of plasma. A typical plasma center, such as those operated by ADMABioCenters, can collect c.30,000L to 50,000L of source plasma annually. This would mean that ADMA would require between 8 and 13 fully operational plasma centers to operate the Boca facility at full capacity. Taking account of this supply dynamic, along with ADMA's current financial position (cash and debt resources), investors should be expecting a slow but steady ramp-up in capacity over the next two years. The terms of the transaction agreement with BTBU, extended a pre-existing RSV plasma supply agreement between ADMA and BPC (Biotest's North American plasma subsidiary). Under this agreement, ADMA has agreed to purchase an annual minimum volume of RSV plasma from BPC. The agreement allows ADMA to supply RSV plasma from up to five ADMABiocenters, and/or to purchase additional RSV plasma from third parties.
The extended agreement is to run for an additional ten years from the close of the transaction. It also covers the supply of hyperimmune plasma to ADMA for the manufacture of Nabi-HB. The terms of the agreement binds Biotest AG and/or its successors (Biotest AG obtains ex-US rights to RI-002 and any new plasma derivatives ADMA develops).
The Boca facility should not experience difficulties in obtaining additional plasma supply agreements. The State of Florida is home to both CSL Ltd.'s global headquarters and CSL Plasma Ltd.'s North American headquarters, which are located just twelve minutes' walk from ADMA's Boca Raton fractionation facility. The state is also home to 39 plasma collection centers (6% of US collection center base) and all of the big six processors maintain at least four collection centers in the state.
8.10: Florida is home to c.40 plasma collection centers
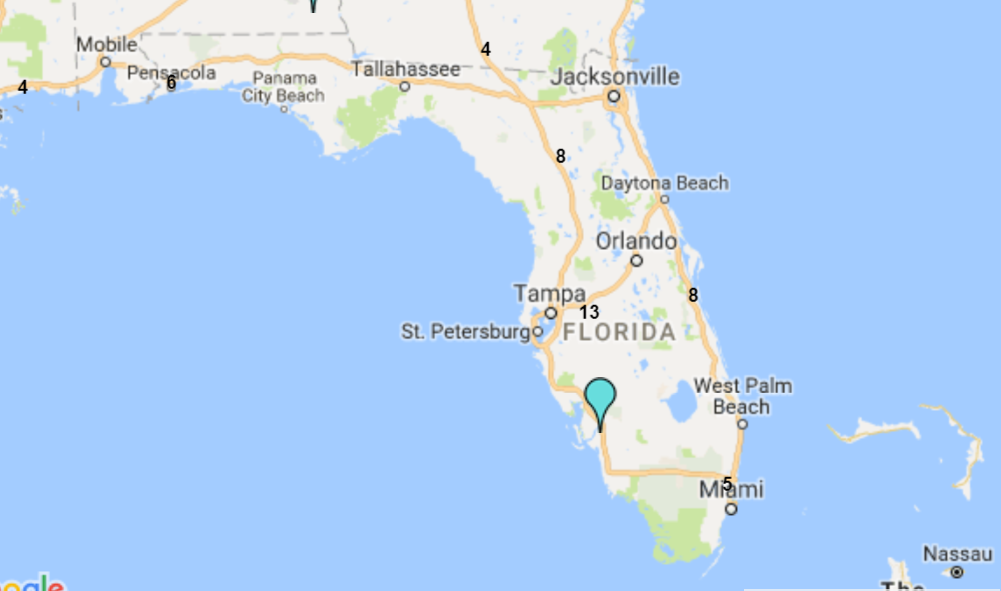
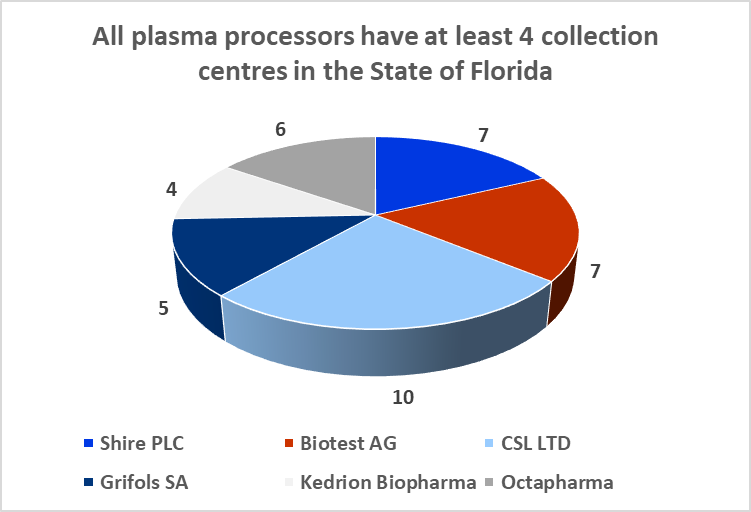
Sources: Donatingplasam.org
9. ADMA can overcome the constraints of last liter economics
The process of fractionating plasma is governed by a set of economic relationships called 'last liter economics' (LLE). The essence of LLE is such that the total potential revenue that can be generated from each gram of protein extracted from each liter of plasma, is gradually reduced from the first liter to the last liter. This means that an economically efficient operation will be attempting to maximize throughput levels, to enhance the volume of extracted proteins (without creating any measurable pathogen contamination risk). The plasma yield is the units of protein/liter of plasma. It is important to note, that while increasing yields is the main focus of the fractionator, a high yield is not necessarily associated with a high potency in terms of the biological action of the protein.
Therefore, the fractionator must also focus on high product quality. Purification processes during the fractionation stage can reduce overall product yields, e.g., when a purification process consists of five steps with yields of 90% each, only 59% of the overall product is recovered. It is therefore of the highest importance that the design of any process incorporates the minimum number of steps, and therefore achieves the highest yield. Under the constraints of LLE, as less and less proteins are produced, each gram must carry an ever-increasing allocation of the plasma and fixed fractionation costs. Fixed costs (i.e., plasma collection) are generally larger than variable costs, which largely increase with volume. This unique cost and revenue relationship has a significant impact on underlying profitability, if the last liter is used to produce one lower value product (such as albumin). The cost structure of fractionating plasma is very different from that seen in similar industrial style manufacturing processes across the wider pharmaceutical sector. Generally, it takes an average of seven to twelve months, from collection of the raw plasma to the completion of the finished product. Working capital is demanding in the plasma derivatives industry and companies carry high inventory levels to comply with regulatory requirements. Government managed reimbursement schemes are major clients of the industry, and this leads to companies holding outsized trade receivables balances. Trade payables tend to be maintained at low levels, as companies pay donors immediately for plasma donations. Some of the large plasma fractionators maintain working capital balances between 50% to 60% of revenues. Grifols, has maintained a level closer to 40%, since it purchased Talecris.
9.1: Impact of LLE on revenue and gross margins at constant plasma and direct fractionation costs
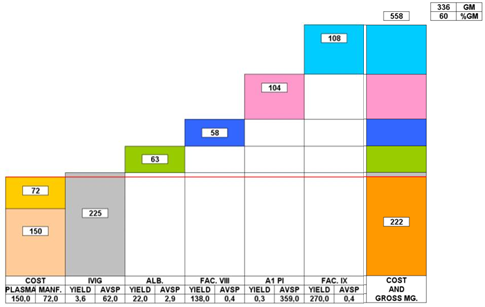
Source: Grifols Presentation
This graph depicts the contribution margin of 5 proteins extracted from a single liter of plasma.
All fractionators use IgG as the revenue driver, but the more efficient processors are more able to cover their plasma and direct fractionation costs from every liter of IgG fractionated. The reality of LLE has pushed the larger processors to achieve greater scale and improve operational efficiencies. The nature of LLE is such that the ability of the fractionator to maximize efficiently gains and create the highest value end products, determines long-term economic success. Creating scale through excess capacity is not an option for the larger processors who have no incentive to use excess capacity if market conditions are not favorable. The economic relationships of LLE can't be altered by the size of the fractionation facility (the fact that facilities less than 100,000L capacity are uneconomical is related to inefficiency and not LLE).
LLE is such that if the ratio of market penetration to volume fractionated is held fixed (% of the market available/liter of plasma fractionated), total potential revenues are the same for a 100,000L facility or a 2,000,000L facility. The composition of plasma is such that only a given number of proteins can be extracted from every liter of plasma. The large processors, fractionate between 4 to 6 proteins, depending on end market conditions. Even if a processor increases the total number of end-products to the maximum number possible, revenues will be higher from the first liter fractionated, but the end-point (where only IVIG and albumin are fractionated) will still be reached and total revenue potential will still be dependent on the total market penetration of the processor and the scale efficiencies it can achieve. If a 4 product versus 6 product market scenario was depicted on a line graph, the shape of the declining revenue line curve would shift depending upon the point in time, when each of the end product markets becomes saturated, making further production uneconomical. As such, it is reasonable to assert that the industry has become less economically attractive for the smaller companies (that lack scale advantages), and even more unattractive for new entrants. The fact that the end market price of IVIG has increased significantly has not changed this dynamic. The increase in price has only changed the baseline of the revenue graph described above. As prices rose, the market has become increasingly difficult for the smaller players, as the larger processors have increased their scale and efficiency advantages.
9.2: A two-scenario revenue model projects a revenue range for the Boca facility of c.$200m-$500m per annum.
To create a firm valuation estimate for the Boca facility, a revenue model is projected using a low-end estimate of the potential revenues that could be generated from a standard 400,000L facility (Cohn/chromatography) processing four standard plasma products (IVIG as the revenue driver), with plasma cost incl. vs. cost not incl). This base estimate shows that a similar fractionation facility could generate c.$200m per annum. The model applies an all-in plasma cost of $150 per liter. It shows that operating profit can be increased by c.45%, when plasma is directly sourced. Management have estimated that a 'back-of-the-envelope'' projection based on current industry guided yields (for IVIG) of c.3.5-4.5 grams per liter (fractionated from a 400,000L facility) gives a potential revenue range of c.$150m to $500m (when the facility is operating at peak capacity). It is important to note, that these are base-level estimates, as that ADMA will be processing higher value hyper-immunes along with standard plasma proteins. This gives ADMA the potential to generate revenues well above these estimates. RI-002 should also be able to produce strong incremental margin growth in comparison to the incremental margin growth potential of traditional plasma products.
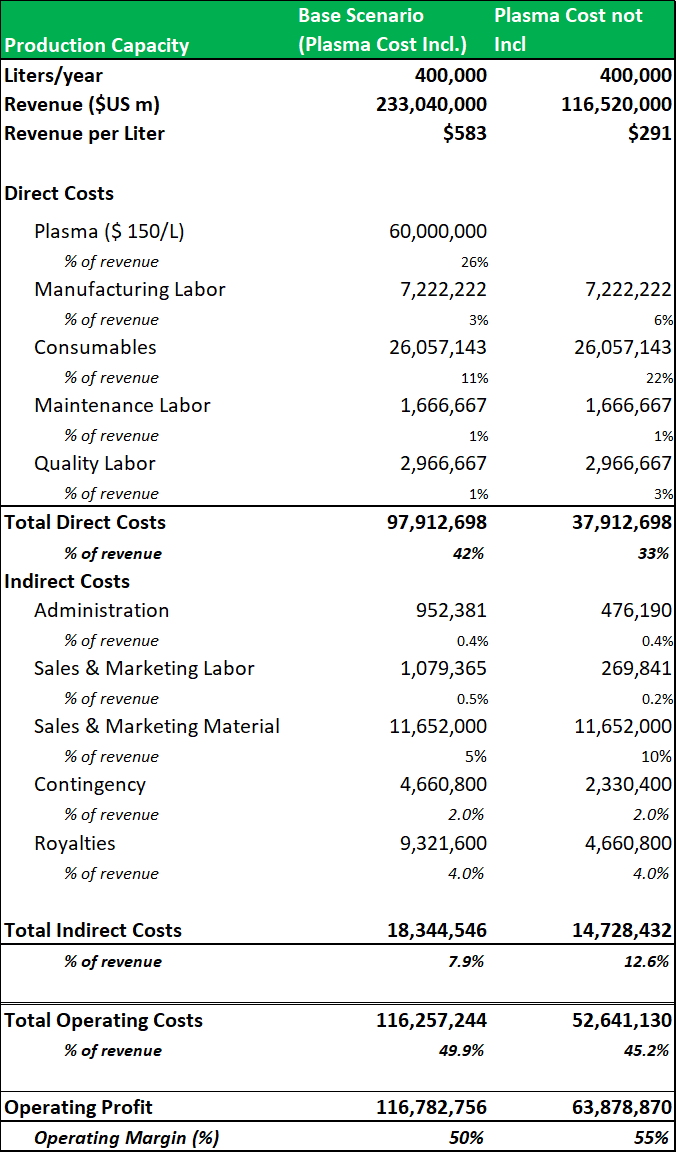
Source: Analyst Estimates
It would not have been economically viable for ADMA to process standard plasma products from the Boca facility with only three collection centers.
Typically, new hyper-immunes products are manufactured in pilot plants (inside the main fractionation facility) and processing typically commences with a throughput range between 1,000L to 5,000L. Another factor in ADMA's favor, is that RI-002's commercial potential won't be limited by a scarcity in plasma supply. Most hyper-immune operations are limited in size by the lack of plasma donors that contain the necessary rare antibodies. This is typically the case for hyper-immune's that have been manufactured for rare viruses such as Ebola, anthrax, rabies, West Nile virus or dengue virus. The patient populations that can benefit from RI-002 treatment, are also larger in size in comparison to these markets.
10. A net asset valuation model for the Boca facility shows that ADMA's shares are trading at a significant discount to fair value
To compensate ADMA for its business losses in relation to the shortcoming at the Boca facility, the BTBU transaction significantly undervalued the assets that were transferred to ADMA. As the land and buildings at the Boca facility are designed for a specialty use, and fair value is too indeterminate, the asset values are written down to a minimal fair value. The transaction placed a total value of less than $20m on the Boca facility itself. Inventory balances, equipment and intangible rights were also transacted at significant discounts to fair value.
Figure 10.1: Value of transferred assets in BTBU Transaction ($m).

Source: ADMA Biologics
Figure 10.2: A NAV model shows that ADMA acquired the facility at a significant discount to fair value.
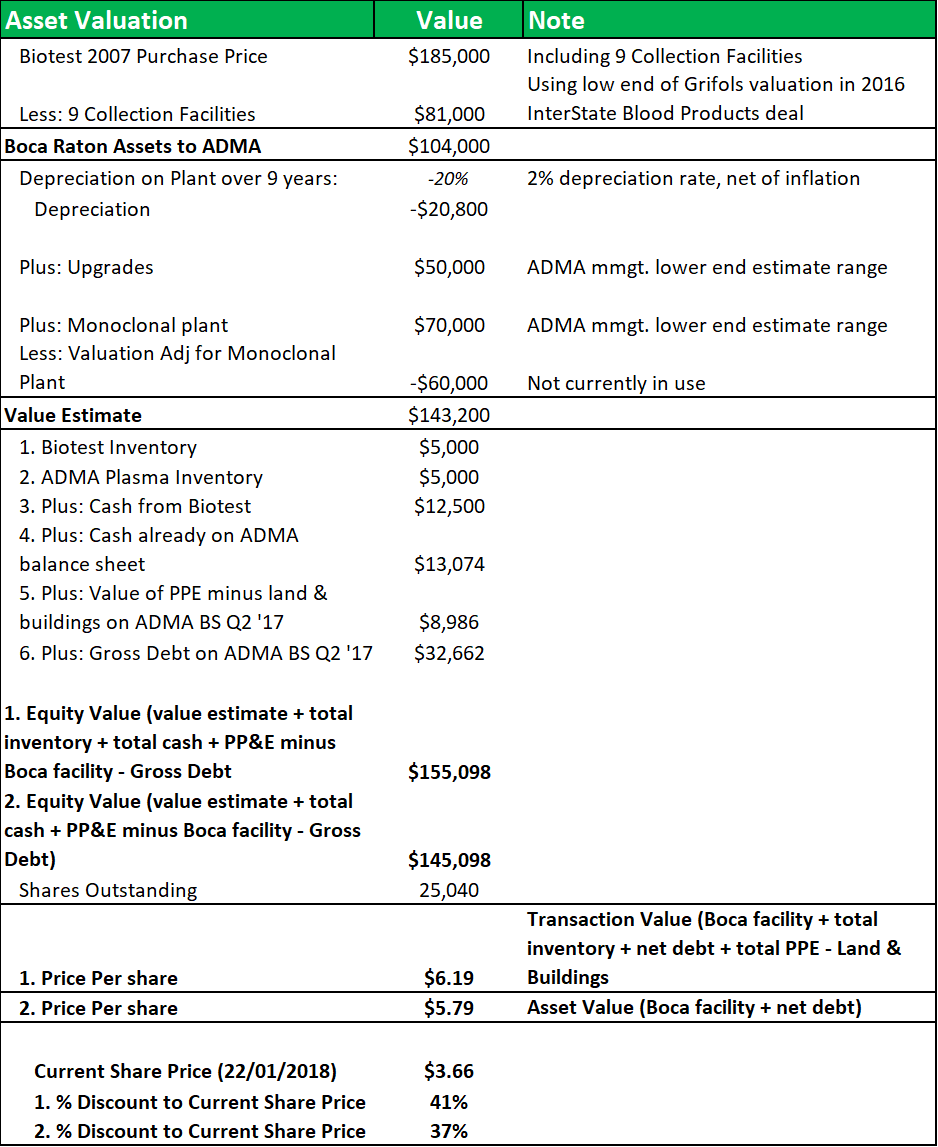
Source: Analyst Estimates
The model breaks down the BTBU transaction by line item and compares the transaction values to the original purchase price that Biotest AG paid for the facility in 2007. Nine collection centers were included in that transaction and their value is subtracted from the calculation at the beginning of the model, using a low-end estimate of the average center values from Grifols' recent transaction with InterState Blood Products. A 2% depreciation rate (net of inflation) is applied. The applicable depreciation rate for the facility, based on a 20-year to 40-year life, was likely between 2.5% to 5% (on a straight-line basis). The value of the land and fixed assets will have appreciated over time, also but depreciated with use. The $50m in capital investment undertaken since 2007, will have offset some of the depreciation. Modeling this range of factors, yields a NAV of between $5.80 and $6.20, or a discount of c.40% to the current share price.
Figure 10.3: The construction of a new 400,000L fractionation facility on a green-field site w/ power, water and other infrastructure already available, would cost in the region of $200m
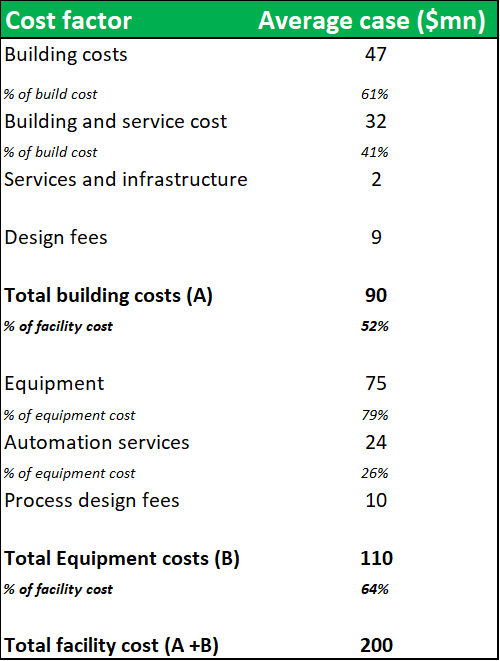
Source: Analyst Estimates
The cost of building a similar facility on a greenfield site, would be greater than $200m. In a transaction scenario, the Boca facility could realize a valuation considerably above these estimates. However, the value of the facility to ADMA's business is much greater than the potential sum of its NAV. ADMA has acquired the facility without needing to incur the high capital investment costs (usually required to build a new facility or expand capacity at an existing facility) and endure the long time periods, needed for construction and obtaining regulatory approval. Only $30.3m of the $63.6m total transaction value was allocated to land, buildings and equipment. Weighting this against the business benefits that ADMA receives through vertical integration, should allow ADMA to unlock substantial value from the transaction. Management has guided that the plant won't require additional capital investment and that Biotest's $50m investment following the 2007 purchase has sufficiently upgraded and automated the fractionation processes (ADMA management say $50m is lower-end estimate). It is possible that this significant NAV discount is due to concerns surrounding the facility's potential illiquidity in a future transaction scenario. This concern is not justified. Although there are only a handful of larger pharmaceuticals currently active in the plasma industry, there are several large private equity firms who have prior experience in the industry. Large fractionation facilities rarely come onto the market, and the opportunity to purchase a large facility, has only been afforded to large firms. Regardless, this thesis does not envision that ADMA will need to liquidate the Boca facility.
Figure 10.4: Recent transactions in the industry display a willingness by acquirers to pay 'full-price' for large facilities
Substantial transaction values have been realized for large fractionation facilities in recent years. CSL Ltd purchased the Rui De plasma business in China for $352m in June 2017 (from Humanwell Healthcare Group). The deal included a large fractionation facility, with a similar peak capacity to the Boca facility. Although the economics of the North American plasma industry are unattractive for new entrants (discussed in section 20 above), there are major growth opportunities in the global plasma industry, particularly in China. The Creat Group, a large Chinese private equity firm (affiliated with Shanghai RAAS, a china-based pharmaceutical conglomerate), recently received approved by CFIUS to purchase Biotest AG for $1.4bn (Biotest had $414.3m in debt). The plasma industry is growing rapidly in China, although fractionation facilities in the country are considered to below international industry standards. Creat Group might be seeking to take advantage of Biotest's recently developed plasma processing facilities in Dreieich, Germany. Creat purchased BioProducts Laboratories (originally created to supply UK national plasma requirements) in May 2016 for £820m (80% owned by Bain Capital and 20% by the UK Department of Health). From conversations with industry representatives it appears that Creat Group, under the stewardship of former BPL CEO John Perkins (formerly of Ampersand Ventures and Talecris) wants to position itself among the top three global plasma processors. It is possible that the firm spoke to Kedrion about a possible deal, before making an offer for Biotest AG. The CFIUS initially blocked the Creat proposal, on potential national security concerns relating to the holding of donor records and related information. A new proposal was accepted in January 2018, with a stipulation that Biotest's North American business is sold to a third party (ownership of these assets is currently held in a US trust). Acquiring Biotest's German technology assets is the most likely justification for the transaction. If the transaction closes, the proposed entity will not have regulatory approval to import plasma into China from the US or Europe, and won't be approved to import plasma from China into Europe. Overall, this activity shows a willingness by large private equity firms to enter an industry that is tightly controlled and largely scale determined. Of course, to compete successfully with the top three companies, the post-transaction entity would need to undertake very substantial capital investment to build the required operational capacity.
Biotest AG was struggling to make inroads against the dominant market positions of the two leading global processors. This dynamic likely played a role in Biotest's decision to recommend that shareholders tender their shares in favor of the Creat bid (and perhaps also in its decision to divest the Boca facility). Biotest had a North American plasma joint venture agreement in place with Kedrion (originally formed in 2014), with the aim of taking market share from the big three processors. These efforts initially delivered top-line sales growth for Biotest, but soon gave way to margin contraction. The BTBU was forced to book large expenses relating to higher unabsorbed manufacturing costs (resulting from the ramp up in staffing levels at the Boca facility) and in relation to unmet anticipated production levels for the distribution agreement (outlined in pg. 89 of Form 14A BTBU transaction proposal).
Biotest's total inventory and asset write-downs for 2016 amounted to €77.2m. The Form 14A transaction proposal, outlines that the BTBU booked inventory provisions totaling $27.6m for FY 2016, while $21.2m was included in the previous year. Some of these amounts were related to the lower of cost to market adjustments and lost validation batches (BTBU failed to pass an FDA inspection required for batch approval). This also demonstrates the disadvantaged strategic positioning of the smaller plasma processors in relation to the big three. Biotest AG probably had a relatively high-cost base, in addition to having a smaller industry market share. In the plasma fractionation business, the extent of the margin pressure experienced when expanding capacity, is determined by the efficiency of the processors cost base.
Figure 10.5: Biotest AG's shares initially rose to an all-time high as investors assessed the company's ability to expand its market share
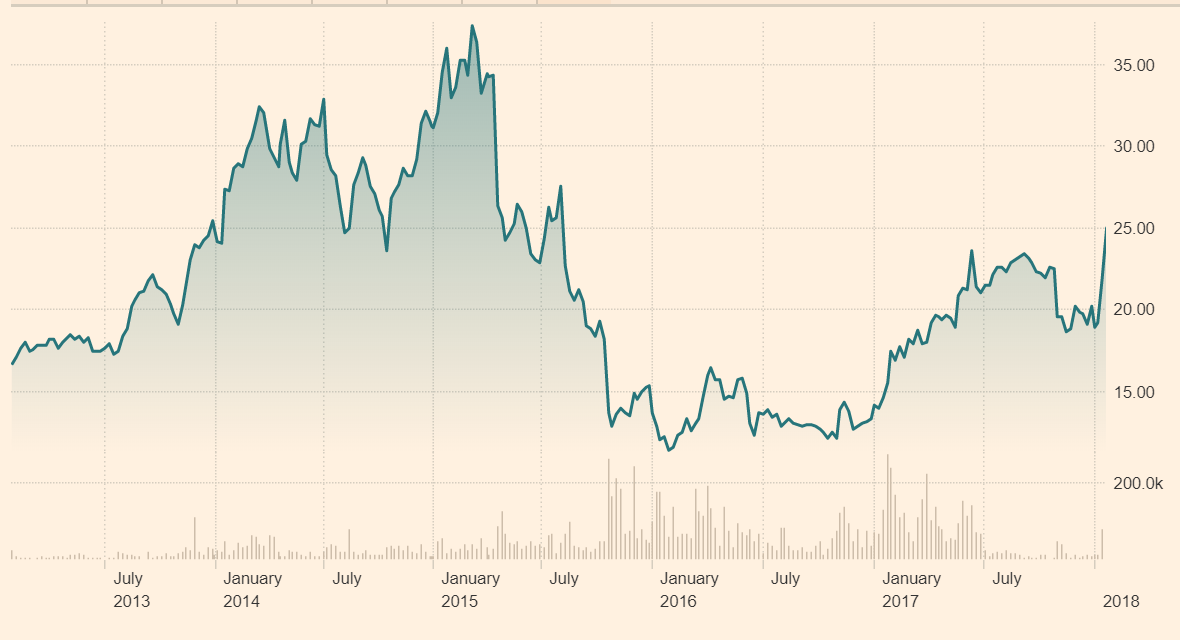
Source: Financial Times
11. "Thou shalt not pass" - ADMA has a business moat that belies its small size
With a wholly owned specially designed fractionation facility, ADMA can now position itself as the sole supplier of a unique high-value hyper-immune product. This niche positioning will allow ADMA to sidestep the industry's extraordinarily high barriers to entry and high degree of market concentration. This was a major point of contention in the 2009 class action lawsuit filed at the Federal Trade Commission (against CSL, Baxter and the PPTA). The filing asserted that the structure of the industry carried a Hirschman/Herfindahl Index score of 2,579 (a score exceeding 1,800 is deemed highly concentrated). The filing also asserted that there was considerable inelasticity of demand in the industry and this was conducive to potential price fixing among the large processors. The lawsuit alleged that the defendants colluded in gathering and communicating market intelligence, which allowed the alleged conspirators to achieve an unfair level of market insight and in-turn fix prices and supply levels. During the hearings, representatives from CSL LTD, admitted that there were "immense barriers to entry" in the industry and there was no realistic prospect for an increase in the number of competing firms. It is reasonable to assert that the market has become even more concentrated since the 2009 trial.
Figure 11.1: The barriers to entry in the plasma industry are four-fold
1. Substantial upfront sunk costs
Each step of the manufacturing process involves substantial up-front, sunk costs; onerous and lengthy regulatory approvals by federal and state agencies; and specialized technical know-how and expertise.
2. Substantial IP portfolio
Market entry requires a significant amount of intellectual property, including trade secrets relating to purification of products and pathogen safety, and substantial product research and development.
3. Extensive capital investment and long lead times
The construction and maintenance of production facilities, including regular improvements necessitated by evolving standards of manufacturing practices, require extensive capital investment and long lead times to obtain the necessary governmental approval. It can take upwards of five years to design, construct and obtain regulatory approval for a green-field plasma fractionation facility. Regulatory hurdles impose significant barriers to entry and extend the time it would take to enter the United States markets, let alone make a significant impact on the market.
4. Necessity of building out large collection center networks
The number of independent plasma suppliers has dwindled significantly, and most independent collection and development capacity is already contracted to existing manufacturers. As such new competitors will need to develop their own domestic-based plasma collection centers and related infrastructure. Only plasma that is collected in the United States is certified for use in products sold domestically.
This combination of competitive factors has made the plasma market very unattractive for new market entrants. In combination with the economics inherent in LLE, a unique competitive dynamic is created in which the only approach to achieving sustainable revenue growth is by increasing production scale and efficiency.
Figure 11.2: Grifols is guiding for a six-year construction and approval process for its new facility at Clayton, North Carolina.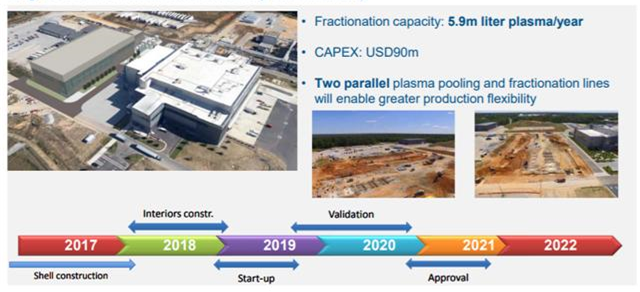
Source: Grifols SA
Achieving economies of scale in plasma collection and fractionation is the main strategic activity for industry participants. The large processors all maintain just one or two very large processing and fractionation campuses in North America. CSL and Grifols in particular, have built up very large processing capabilities in North America which has given them near insurmountable barriers to competition.
Alongside the lengthy construction and approval time frames mentioned above, there is the added complexity involved in either developing in-house engineering expertise or in finding third party contractors (very few engineering companies have expertise in large specialized projects in the pharmaceutical space). Grifols SA maintains its own in-house engineering division for large projects. In February 2016, its Board of Directors approved a new $360m capital investment plan specifically focused on its plasma manufacturing operations, covering the period 2016-2023.
It plans to develop 5.9m liters of additional fractionation capacity at its main fractionation center in Clayton, North Carolina (expected capital outlay of $90m). Grifols is guiding for a six-year construction and approval process at the facility. CSL LTD doesn't disclose specific details on its capital investment projects, but the company has invested $2.6bn in projects over the last five years, and has guided for a future capital investment rate of between 10% and 11% of sales.
Figure 11.3: Along with the industry-wide barriers mentioned above, AMDA's business will have several unique barriers
1. The regulatory regime, is a significant barrier to entry
The structure of the regulatory regime covering the development, production and commercialization of plasma treatments will be a major deterrent for potential competitors. In addition, ADMA's products are protected by intellectual property rights and trade secrecy laws.
2. ADMA has patents and trade secrecy protections
The criteria and methodologies that ADMA uses to select its plasma donors, formulate its plasma pools, and validate its micro-neutralization assays (for anti-RSV antibody detection in human plasma and in the final product) are patented. ADMA's collection of patents and trade secrets cover unique and exclusive reagents, controls, reference standards and methods used in the assay process.
3. Significant investment would be required to replicate ADMA's specially designed hyper-immune facility
The Boca facility is operated to a high degree of technical complexity, and almost all aspects of the fractionation process are validated by the FDA. The construction of a new fractionation facility will require significant investment.
4. ADMA has built up considerable technical and scientific know-how while developing RI-002
To commercialize a competing therapy, competitors will need to invest substantial resources in developing the required technical and scientific know-how, before undertaking a long development program. The FDA requires that all new IgG products are developed as first in class treatments. This protects existing therapies from generic competition (which can reduce average sales prices by up to 90% in the wider pharmaceutical sector).
12. ADMA's management team have surrounded themselves with experienced industry executives
ADMA was originally founded in 2004 by Adam Grossman and his father Dr. Jerrold Grossman. The father and son team obtained their experience of the plasma industry with companies such as Genesis BioPharmaceuticals, National Hospital Specialities and Medimmune. The CEO Mr. Adam Grossman was a sales and business development executive at Medimmune during the commercialization of Synagis. Now that RI-002 has passed through a protracted (and costly) development process, management are more motivated than ever to succeed in their goals. To oversee operations at the newly acquired facility Mr. Grossman has relocated with his family to Boca Raton, Florida. In addition, ADMA's Board of Directors includes leading experts from the fields of healthcare investing, venture capital, and the plasma industry (collectively, management and the board of directors have over 150 years of substantial medical, technical and development experience across the industry).The board includes Mr. Laurence Guiheen, the COO of Kedrion (and formerly a president of Baxter BioSciences), who has experience in plasma fractionation and has served on board of the PPTA. The board also includes, Dr. Bernard Ehmer the CEO of Biotest AG, who currently sits on the global Board of Directors of the PPTA.
13. RI-022 can reach over $200m in revenues within five years of market entry.
Market model assumptions
- A peak market penetration rate among PIDD patients of 33%.
- Average patient weight of 46kg (taking account of average adult and infant weights).
- Average cost price of $450 per gram, at a dosage volume of 27g per dose, with average cost price inflation of 2.3% over the period.
Figure 13.1: With a peak penetration rate of 33% in the PIDD patient segment, RI-002 can generate over $50m in revenues within five years of market entry


Source: Analyst Estimates
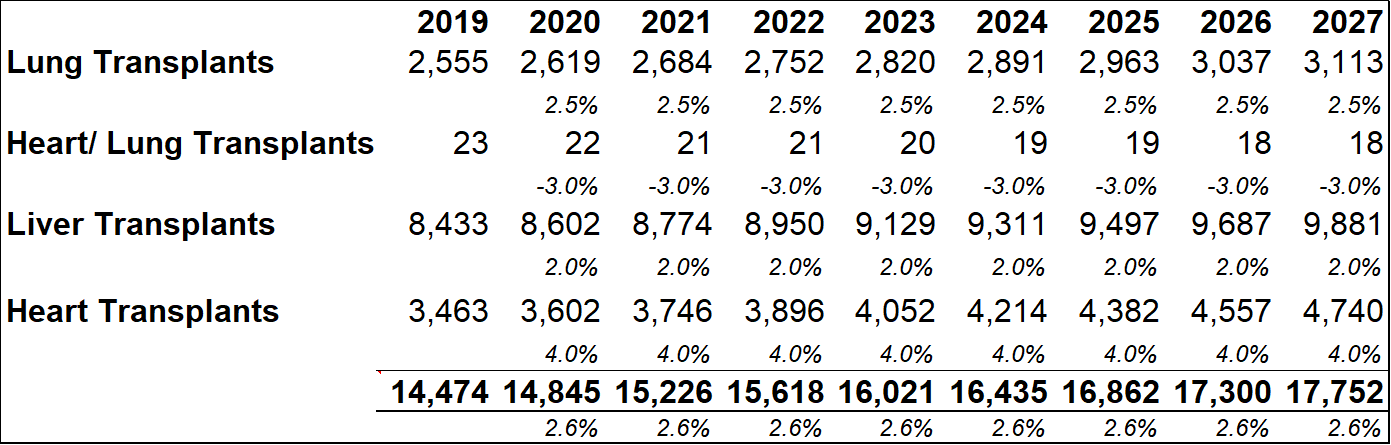
Important points to note, in relation to average dosage volumes, are that most patients require minor differences in total dosage volumes depending on each patient's immune physiological differences and comorbidities. The AAAAI guidelines recommend that patients with PIDD are treated every three to four weeks with doses ranging between 400 mg/kg to 600 mg/kg. Currently there is no defined optimal dose for IG replacement therapy even though the treatment has been in use for more than 50 years. The model projects that within five years of market entry, RI-002 can be treating c. 2,000 PIDD patients per year (generating between $80m and $90m in revenues). As discussed above patients undergoing solid organ transplant operations (SOT's) can benefit from treatment with RI-002. The table below (figure 23.2) outlines future projections based on historical data obtained from the Organ Procurement and Transplant Network. While these figures could fluctuate somewhat, the projected yearly total figures provide a reasonable guide for this model. The model projects total future transplant numbers for each of the relevant transplant sub-groups, based on historical 10-year average growth or decline rates for each sub group.
Figure 13.2: Collectively, the four transplant groups that can benefit from RI-002 use are projected to grow by approximately 2.6% y-o-y
Source: Analyst Estimates
Figure 13.3: The SOT revenue model uses conservative penetration estimates to account for the lack of detailed patient data.
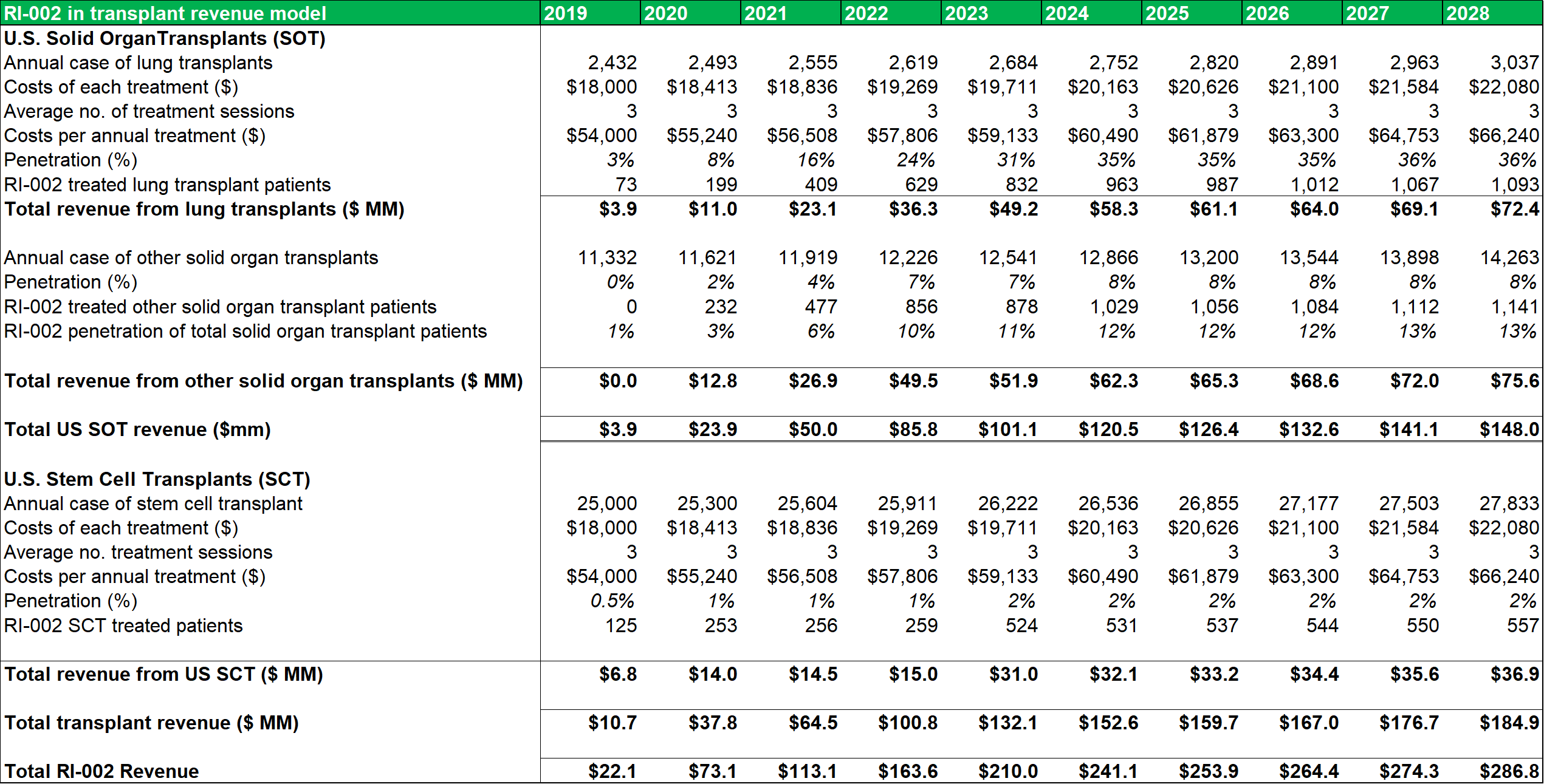
Source: Analyst Estimates
Among the transplant subgroups, patients undergoing a lung transplant (who contract RSV and/or are at a heightened risk of contracting RSV due to IC status) might benefit the most from RI-002 treatment. As such, the model assumes a peak penetration rate of 33%, with a penetration rate of 31% being reached within five years. A more conservative rate of 12%-13% is modelled for other SOT's. As discussed above, patients who become immune compromised while undergoing stem cell transplants, can benefit from RI-002 use (to counteract/prevent RSV infection). To account for the difficulty in estimating potential incidence rates in this subgroup, a very conservative 2% peak penetration rate is used.
Figure 13.4: Post transaction ADMA can generate a blended gross profit margin of c.60% and a blended operating margin of c.45%.
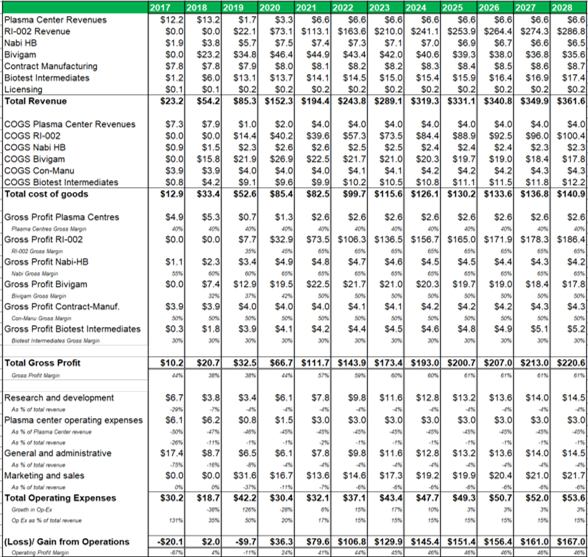
Source: Analyst Estimates
The margin analysis model (figure 23.4) uses management guidance for top-line and bottom-line margins and projects COGS, gross profit and operating expense margins for each business segment. The plasma collection centers are projected to generate a 40% gross margin, while RI-002 (now fractionated from ADMA's wholly owned fractionation facility) can generate a gross margin of 65% after five years of revenues. Marketing and sales is projected to account for 6% of revenues, research and development is projected to account for 4% of revenues, while general and administrative expenses are expected to account for 4% of total revenues (all after five years of RI-002 revenues).
Figure 13.5: A risk-adjusted net present valuation model yields a fair value of $15.17 for ADMA shares.
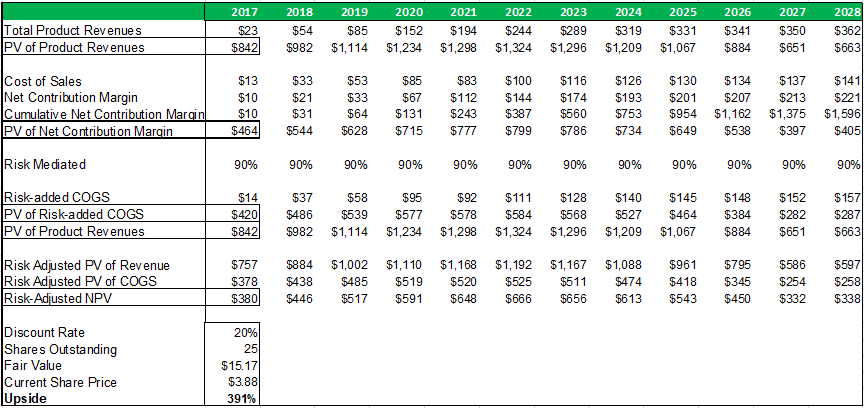
Source: Analyst Estimates
Taking the present value of ADMA's total projected product revenues (from all business segments), along with the present value of all projected COGS expenses (from all business segments), and risk adjusting both for a 90% percentage of success (POS), a total risk adjusted net present value of $380m is reached. The 90% POS adjustment takes account of RI-002's stage of development, and the high likelihood that the Boca facility will pass a cGMP re-inspection and the BLA for RI-002 can be refiled.
Figure 13.6: A P/E valuation model applying a 12x P/E multiple to2020E EPS and 2021E EPS, yields a fair value of $16.35 for ADMA shares.
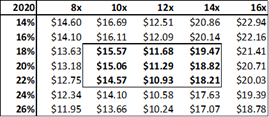
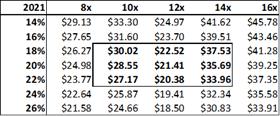
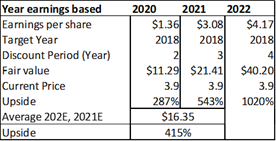
Source: Analyst Estimates
The P/E valuation model uses a blended fair value based on 2020E EPS and 2021E EPS to account for RI-002's strong EPS growth over the period (2020E to 2021E projected top-line revenue growth is 28% versus EPS growth for the same period of 128%). A 12x P/E multiple is a conservative valuation multiple for ADMA, considering its potential to achieve niche positioning. ADMA's peers in the plasma industry have conglomerate business models and global operations, and therefore attract premium P/E multiples in the range of 20x to 30x. While ADMA's will be a member of this peer group it won't be competing directly against these companies. Emergent BioSolutions (Ticker: EBS US Equity), a publicly listed Canadian company that specializes in vaccines for rare diseases and infections, is perhaps a more directly comparable company (for valuation purposes). Like ADMA, this company has a niche market focus in a highly technical industry. This company's unique competitive positioning creates a substantial moat around its business. As a result, its shares attract a premium valuation, currently trading at a P/E of 28x, and a P/S of 4x. The company has a market capitalization of $2.4bn, and generated EBITDA of $136.9m and EBIT of $98.6m for FY 2017. When ADMA has built up a strong revenue base from RI-002 sales, it could attract a premium valuation, and its shares could trade with a P/E multiple in the range of 20x to 25x.
Figure 13.7: A technology valuation model comprised of 10 randomly selected biotechnology companies demonstrates the overly-severe valuation discount currently placed on ADMA's patented technology
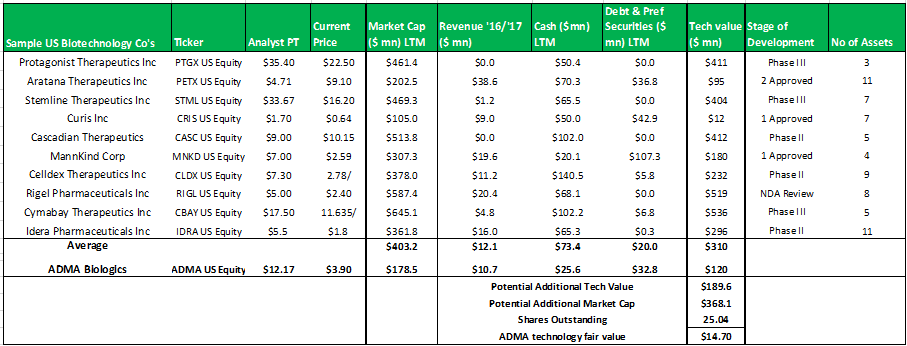
Source: Analyst Estimates
The model above, selects a random group of ten publicly traded biotechnology companies that have at least three assets in development (some have assets that have received regulatory approval). The model subtracts the cash and debts values from the market capitalizations to derive the implied technology values for each company. The group average technology value is added to ADMA's current market capitalization to derive a fair value for ADMA's technology of $14.70. When considering the strength of this valuation estimate it is important to remember that the stock market often misprices biotechnology assets.
In summary, an average of the rNPV fair value of $15.17 and the blended 12x P/E fair value of $16.35, yields a fair value for ADMA shares of $15.76. Applying risk adjusted fair value valuation methodologies for ADMA's near-term future potential earnings, is perhaps the best approach for deriving a fair value for ADMA's shares given the unique situation the company has found itself in. ADMA is a very attractive investment from a value perspective as the shares present a very favorable margin of safety. ADMA's current enterprise value of $185.7m, is considerably less than the fair value NPV of the Boca facility (in and of itself, w/ no working capital or inventory balances).
14. ADMA's shares offer a unique opportunity for the long term investor.
Summary of the investment case
- ADMA's lead product candidate has generated very positive clinical data, and is positioned to meet a very high unmet medical need. ADMA has received a CRL that didn't cite issues with the drugs clinical profile. The scientific profile of IVIG treatments in immune compromised patients is very developed, and RI-002's completed clinical program is more than sufficient to obtain approval.
- ADMA has acquired a specialized hyper-immune fractionation facility, without having to expend high upfront capital investment costs or pass a burdensome approval timeline. ADMA will be the first company in the plasma industry to fractionate a high-value hyper-immune from a 400,000L facility. This gives the company a niche position within the highly entrenched plasma industry and can allow it to achieve ' premium-product' profit margins, while being protected with a significant business moat.
- The burden of lifting the regulatory hold at the Boca facility is much lower than might be perceived by investors at present. The regulatory landscape pertaining to fractionation facilities is very well defined by the various regulatory bodies and ADMA has obtained the services of an experienced consulting firm.
Summary of the investment risks
- Management could be underestimating the need for additional capital investment at the Boca facility. Management have guided that the facility is in very good condition, and that the $50m capital investment estimate by BTBU likely underestimates the total amount invested. The CRL did not cite the need for the facility to be physically upgraded but instead focused on processes and employee training and skills.
- Managing the Boca facility will be a challenging task for ADMA's management. Although, AMDA's executives have significant experience in the plasma industry, they have relatively little direct experience in managing a fractionation facility. In addition, it will take several years before capacity can be fully scaled and a large number of additional employees will need to be hired. Also, AMDA intends to commercialize RI-002 without engaging third parties. There is a risk that management are underestimating the operationally burdensome nature of operating a large fractionation facility and simultaneously commercializing a new product candidate.
- Management have guided that its current liquidity and cash resources will be sufficient to see the company into the second half of 2018, when its revenues should increase considerably. If AMDA was to fail the FDA re-inspection, timelines for its major business objectives will be pushed back into the future. This would place significant strain on the company's balance sheet. As additional capital would be required, significant dilution for current shareholders could result.
To operate the facility efficiently, ADMA will need a steady supply of high quality plasma. Two of its existing collection centers will transfer to Biotest at year-end 2019 and a third center has only recently been established. Although additional supply agreements are already in place, ADMA's supply partner could experience business disruptions in its supply chain which could deprive ADMA of timely supplies of raw plasma. Although the state of Florida is a hub for the plasma industry in the South-Eastern United States, and all of the main plasma processors maintain at least four centers in the state, these companies could decline to supply ADMA in the event of supply shortages in the industry.
ADMA's shares offer a unique opportunity for long-term investors. It is very rare for highly specialized plasma facilities to come onto the market and ADMA has been afforded a unique opportunity to acquire a very valuable asset. When viewed in the wider context of very richly valued asset markets, it is extraordinary that ADMA's key asset is trading well below replacement value. ADMA is pursuing important business objectives, which have the potential impact to impact patient outcomes. It will be first company in the industry to manufacture a polyclonal plasma derived hyper immune. Serum antibodies recovered from convalescent patients have been in use in the medical field since the 1900's (origins of immunotherapy). Although the technology's in use in the industry have greatly increased efficiency, the end market need for the plasma derivatives has continued to grow unabated. For investors with one eye on the business cycle, it is important to remember that the plasma industry was not overly affected by the 2008 GFC.
INCOME STATEMENT
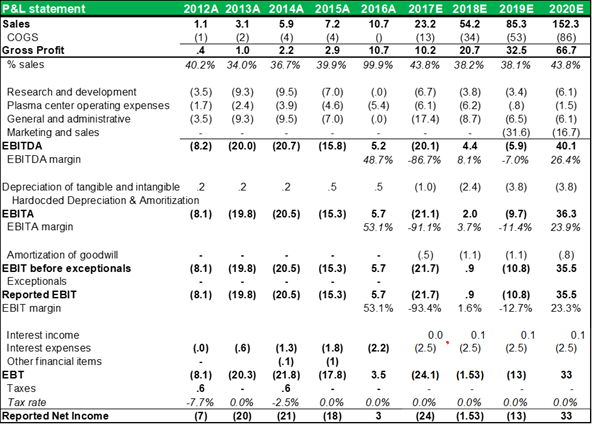
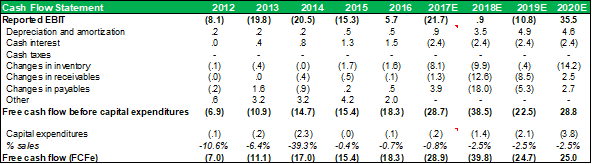
CASHFLOW STATEMENT
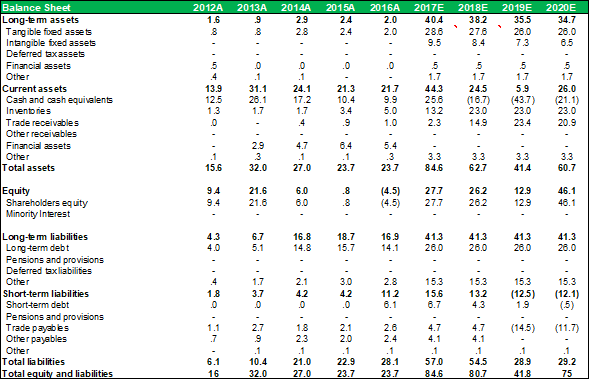
BALANCE SHEET
Important: Disclaimer
No aspect of this research piece is intended to be relied on as investment advice. The content is intended to be used and must be used for informational purposes only. It is very important to do your own analysis before making any investment based on your own personal circumstances. You should take independent financial advice from a professional in connection with, or independently research and verify, any information that you have read in this report and wish to rely upon, whether for the purpose of making an investment decision or otherwise.
Appendix
1.1 Comparable's tables
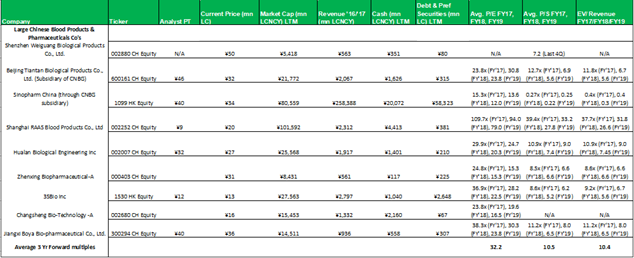
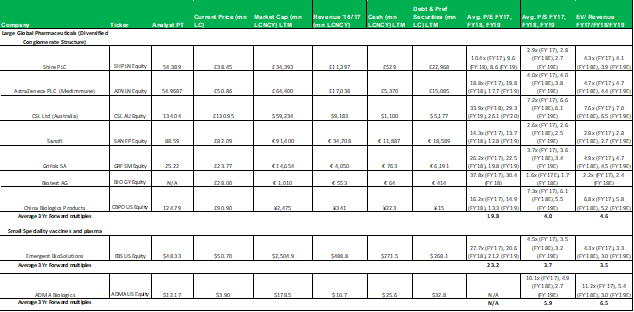
No aspect of this research piece is intended to be relied on as investment advice. The content is intended to be used and must be used for informational purposes only. It is very important to do your ...
more